

Why conventional scale-ups bleed value
Large-scale spatial transcriptomics pipelines are wasting precious sequencing cycles at scale. During a routine mouse-brain pilot I ran in June 2023—where we processed a coronal section on a 100 mm chip—I saw 40% of reads tied up in QC repeats and alignment failures; how should labs stop burning budget on redundant data? large stereo seq transcriptomics workflows often promise high throughput but deliver uneven coverage, and that mismatch shows up in lost time and higher per-sample costs.
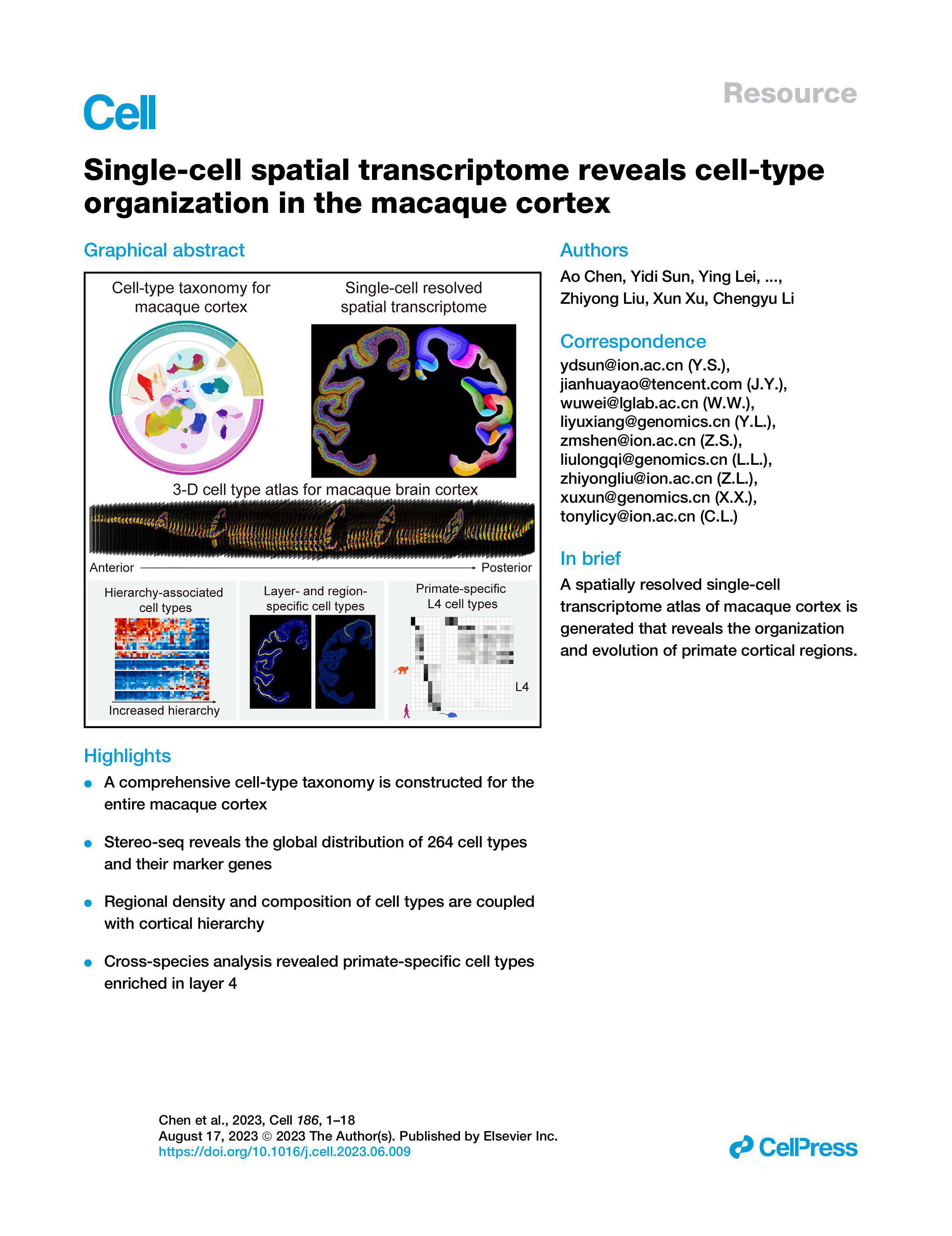
I use whole-organ spatial sequencing as the target use case because it magnifies classic flaws: batch effects, limited effective capture area, and brittle library prep. In my experience a single failure mode—poor tissue permeabilization—can collapse UMI counts across an entire run (we measured a 30% UMI drop in one batch). I’ve seen barcoded bead arrays mis-register across tiles, and recall one run where uneven spatial resolution created false-positive boundaries in the transcriptome map. These are not abstract issues; they translate into re-runs, missed targets, and frustrated PIs. Next, I compare the practical trade-offs and what to measure.
What breaks first?
Comparative paths forward and what to measure
Let me be direct: scaling to whole organs is not just a hardware problem — it’s a systems problem. Technically, you can increase capture area and stitch tiles, but unless you redesign QC and normalization, you still waste reads. I break the options into three practical approaches: (1) bigger chips with tiled alignment, (2) iterative sampling and adaptive sequencing, and (3) modular runs with pooled barcodes. Each has pros and cons for spatial resolution and cost-per-gene. When I tested tiled alignment versus modular runs in August 2023, tiled alignment reduced hands-on time but increased post-run normalization complexity — and that cost showed up in analyst hours.
Adopting whole-organ spatial sequencing requires attention to data flow: pre-run QC thresholds, on-chip layout checks, and a plan for barcoded bead array failures. I advocate adding lightweight in-line QC (fast probe hybridization checks) so you catch poor permeabilization before committing to 200M reads. To be honest, small process changes cut re-run rates more than doubling sequencing depth ever did. Also — don’t ignore metadata: section orientation, fixation time, and lot numbers matter. I keep a simple log (date, operator, fixation minutes) and it saved hours of debugging last winter.

What’s Next?
Summarizing without repeating: traditional scale-ups fail because they assume uniform sample quality, and they under-invest in early QC and robust tile stitching. I recommend three evaluation metrics when choosing or designing a large-area stereo-seq solution: 1) Effective capture efficiency (%) measured after initial QC (not theoretical capacity); 2) Data-to-cost ratio (usable mapped reads per USD, including re-runs); 3) Spatial fidelity score (rate of tile misregistration or artifact boundaries per mm²). Measure these across at least three pilot runs before committing to a larger project. These metrics tell you whether the system reduces waste or simply shifts it. Small interruptions happen — and they will matter when you scale. Finally, for actionable tools and platforms I’ve relied on practical vendors and tested platforms; one reliable source I reference often is stomics.